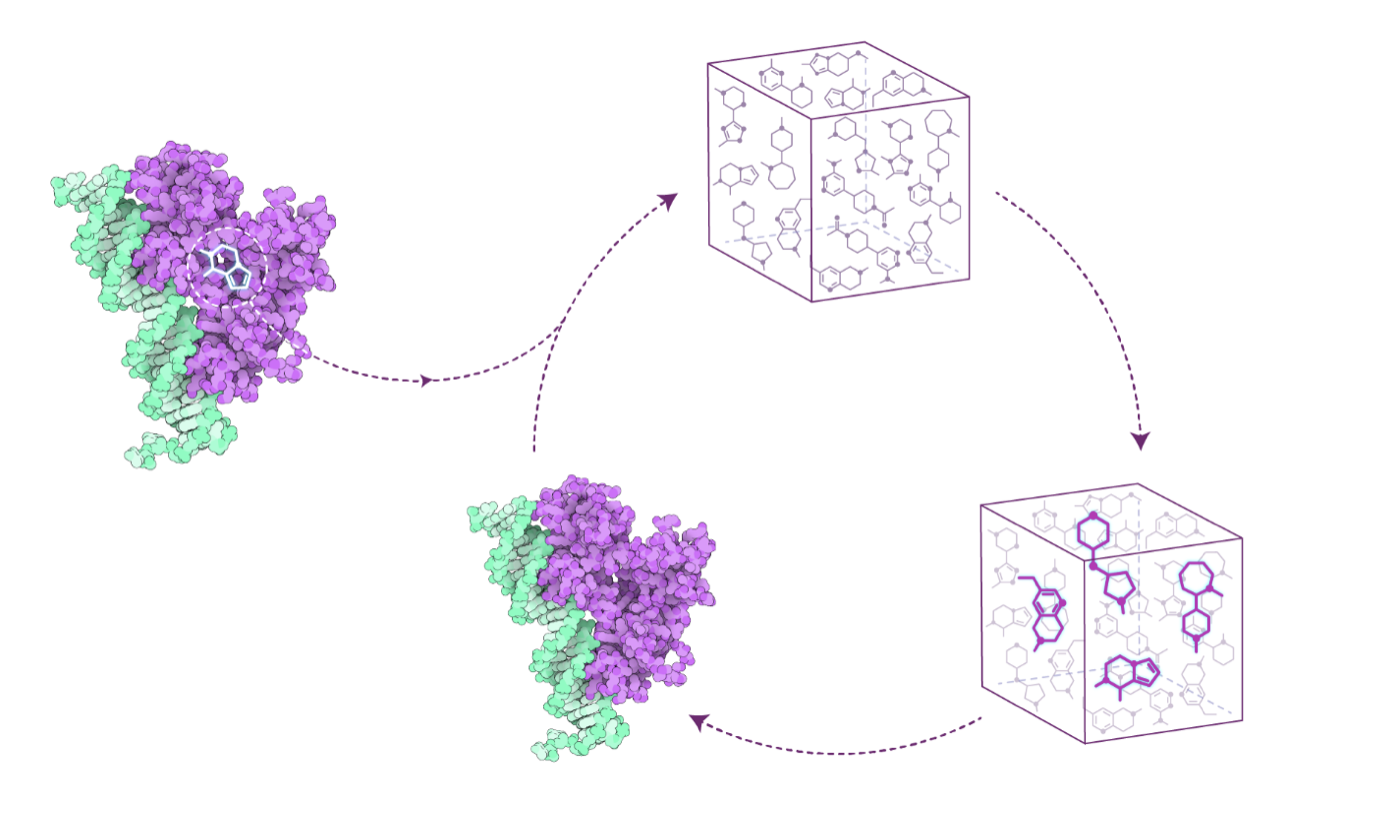
What it is
Mexa Design is our AI-powered platform that creates cyclic peptide drug candidates from scratch — even when no experimental leads exist. It turns the toughest starting point in drug discovery into a concrete set of optimized candidates.
Why it’s different
- In silico first – Structure-based rational drug design, not random screening.
- AI-enhanced chemical space – Beyond traditional docking, our AI expands the cyclic peptide universe to include non-canonical amino acids.
- Synthesizable candidates – Algorithms are tuned to ensure amino acids are available for real-world synthesis.
- Customizable – Add up to five of your own non-canonical amino acids to the design pool.
Who it’s for
- Researchers with new biological targets but no starting chemistry.
- Teams interested in exploring cyclic peptide modalities.
- Projects focusing on competitive inhibition, receptor inhibition, radiopharmaceutical targets, or protein–protein interfaces.
What you get
- 15 cyclic peptide drug candidates, optimized for binding affinity and drug-like properties.
FAQ Highlights
- Is this in silico design? – Yes. Structure-based design combined with AI-generated chemical space.
- Why trust the results? – High-accuracy computational methods, validated workflows, and synthetic feasibility checks.
- Are candidates closer to drugs? – They are optimized starting points. Binding is central, and optimized for drug-like properties. To obtain drug candidates, project-based development is required.
- Can you add my amino acid? – Yes. We can integrate your custom amino acids into the design pool.